
医疗器械产品设计流程
-

-
xbs999 这家伙很懒,还没有设置简介
0 人点赞了该文章 · 953 浏览
一. 设计和开发总则
研发合规成为监管热点,设计开发过程管控尤为重要,其设计开发主要依据如下标准;
ISO13485的第7.3设计开发
FDA 21CFR820 820.30设计控制
中国GMP的第六章设计开发
注册质量管理体系现场核查指南
注意:最晚从注册送检样品开始必须完全符合体系要求!
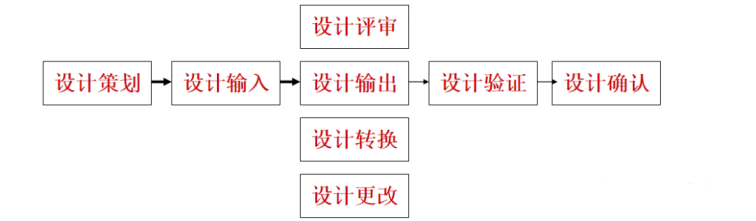
每个阶段相关详细内容如下:
1.1设计和开发策划
组织应策划和控制产品的设计和开发。适当时,随着设计和开发的进展,应保持和更新设计和开发计划文件。
(1)设计和开发阶段
(2)每个设计和开发阶段所需要的评审
(3)适用于每个设计和开发阶段的验证、确认和设计转换活动
(4)设计和开发的职责权限
(5)为确保设计和开发输出到设计和开发输入可追溯性的方法
(6)包括必要的人员能力在内的所需资源
1.2设计和开发输入
应确定与产品要求相关的输入并保持记录,这些输入包括:
(1)依据预期用途,功能、性能、可用性和安全要求;
(2)适用的法规要求和标准
(3)适用的风险管理输出
(4)适当时,以前类似设计提供的信息
(5)产品和过程的设计和开发所必需的其他要求
应当对这些输入的充分性和适宜性进行评审和批准。要求应完整、明确,能被验证和确认,并且不能自相矛盾
1.3设计和开发输出
设计和开发输出应
(1)满足设计和开发输入的要求
(2)给出采购、生产和服务提供适当的信息
(3)包含或引用产品接受准则
(4)规定对产品的安全和正常使用所必需的产品特性
设计和开发输出的形式应适合于设计和开发输入的验证,并应在发布前批准,应保持设计和开发输出的记录
1.4设计和开发评审
在适宜的阶段,应依据策划和文件化的安排,对设计和开发进行系统的评审,以便:
(1)评价设计和开发的结果满足要求的能力
(2)识别和提出必要的措施
评审的参加者应包括与所评审的设计和开发阶段有关的职能的代表和其他的专家。
评审结果及任何必要的措施的记录应予保持
1.5设计和开发验证
为确保设计和开发输出满足设计开发输入的要求,应依据所策划和文件化的安排对设计和开发进行验证。组织应将验证计划形成文件,包括方法、接受准则,适当时,为确定抽样量所采用的统计技术与原理。
如果预期用途需要医疗器械与其他医疗器械连接或接合时,证实设计输出满足设计输入的内容。
验证结果和结论以及必要措施的记录应予保持。
1.6设计和开发确认
为确保产品能够满足规定的适用要求或预期用途的要求,应依据所策划并文件化的安排对设计和开发进行确认。
组织应确认计划形成文件,包括方法、接受准则,适当时,未确定抽样量所采用的统计技术和原理。
应对代表性产品进行设计确认,代表性产品包括最初的生产单位、批或其他等同物。应记录用于进行确认的产品的合理性。
作为设计和开发确认的一部分,组织应按照适用的法规要求进行临床评价或性能评价。
用于临床评价或性能评价的医疗器械不应视作放行给顾客使用。
如果预期用途需要医疗器械与其他医疗器械连接或接合,确认应包含依此连接或接合时,证实规定的适用要求或预期用途已得到满足的内容。
确认应在产品交付给客户使用之前完成。
确认结果及必要措施的记录应予保持
1.7设计和开发转换
组织应将设计和开发输出到制造的转换程序形成文件。这些程序应确保设计和开发的输出在成为最终生产规范之前以适用于生产的方式经过验证,并且生产能力能满足产品要求。
转换的结果和结论应予记录
1.8设计和开发变更
组织应将控制设计和开发变更的程序形成文件。组织应确定与医疗器械的功能、性能、可用性、安全和适用的医疗器械法规要求和其预期使用有关的重要变更。
(1)经过评审
(2)经过验证
(3)适当时,经确认;
(4)经过批准
设计和开发变更的评审应包括过程中或已经配送的部件和产品的变化和风险管理和产品实现过程的输入和输出的变化的影响的评价,
更改的评审结果及任何必要措施的记录应予保持
设计和开发更改的评审应包括评价更改对产品组成部分和在制品或已交付产品的影响,评价更改对风险管理的输入/输出和产品实现的过程的影响。
1.9设计和开发文件
组织应保持每一医疗器械类型或医疗器械族的设计和开发文件,此文件应包括或引用为证实符合设计和开发要求所产生的记录,以及设计和开发变更的记录。
二. 设计和开发流程
设计和开发流程总图,详见图表;
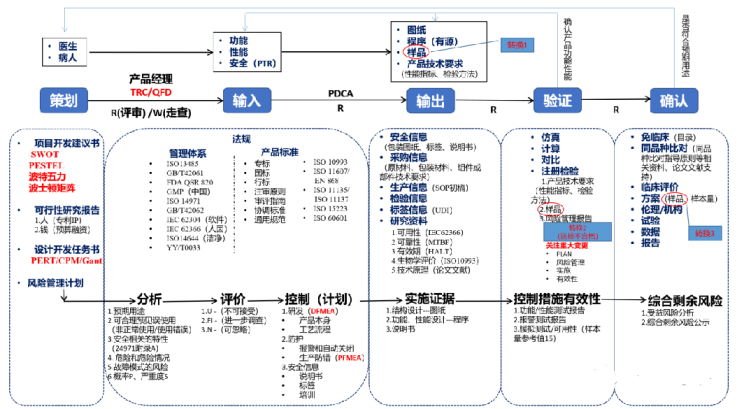
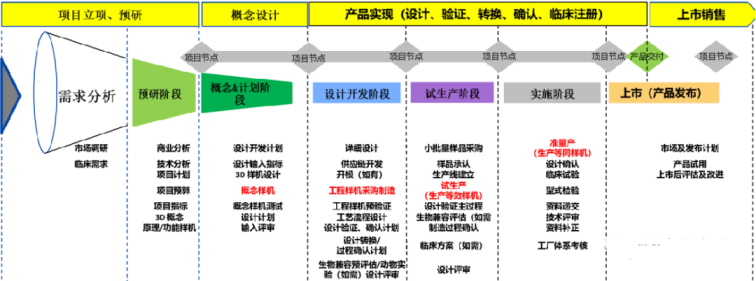
三. 产品注册与质量管理体系核查
产品注册与质量管理体系核查的过程和节点,如下图所示
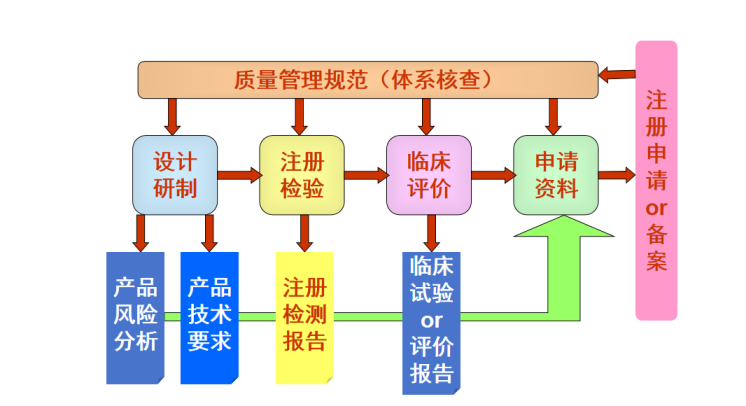
****************************************
全部 0条评论