
知识分享|基于风险的思维来管理不合格和偏差
-

-
多多猪 这家伙很懒,还没有设置简介
0 人点赞了该文章 · 5502 浏览
Probably the most significant concern for anyone responsible for implementing, deploying, and maintaining a quality management system (QMS) is the integration of risk-based thinking. While the concepts of risk-based thinking and management are not new, previous practice was more reactionary, primarily focusing on detection after the fact, root cause analysis, corrective actions, and preventing recurrence of the failure. Contemporary thinking places the emphasis on considering risks up front (prevention) and having a solid approach to address risk in planning, managing, and driving actions.
对于负责实施、部署和维护质量管理体系(QMS)的人员来说,风险思维的整合是一个重要的问题。虽然基于风险的思维和管理并不是最新提出的概念,但是以前对风险思维的应用主要集中在事后的检测、根本原因分析(root cause analysis)、纠正措施(corrective actions)以及防止失败再次发生方面。当前的风险思维则将重点放在预防风险上,强调在计划、管理和执行等全过程中设定解决和预防风险的方法。
This article presents the requirements regarding nonconformances and deviations, and then introduces some tools to incorporate and integrate risk management techniques within the QMS, specifically applied to nonconformance and deviation management.
本文介绍了有关不合格项(Nonconformances)和偏差(Deviations)的规定,然后介绍了可纳入质量管理体系中的适用于不合格和偏差管理的风险管理技术工具。
一、Requirements And Background 要求和背景
There several International Organization for Standardization (ISO) standards, Food and Drug Administration (FDA) regulations, and national and international guidance documents that provide direction and lay out the framework for successfully implementing, maintaining, and sustaining an effective and robust quality management system. The standards, regulations, and guidances require the management of nonconformances and deviations for products and services provided. Risk-based thinking can help prioritize nonconformance and deviation management. The applicable standards, regulations, and guidances include, but are not limited to, the following:
国际标准化组织(ISO)的标准、美国食品药品监督管理局(FDA)的规定,以及各国和国际间的指南文件为成功实施、维持有效和健全的质量管理体系提供了指导和框架。相关标准、法规和指南文件中明确提出要对产品或服务的不合格项和偏差进行管理。而基于风险的思维可以帮助在不合格项和偏差出现之前进行管理。相关的适用标准、规定和指南文件包括但不限于以下内容:
ISO 9001:2015—Quality management systems — Requirements 质量管理体系—要求
8.7.1 The organization shall ensure that outputs that do not conform to their requirements are identified and controlled to prevent their unintended use or delivery.
8.7.1 组织应确保识别和控制不符合要求的输出(outputs),以防止其被非预期使用或交付。
The organization shall take appropriate action based on the nature of the nonconformity and its effect on the conformity of products and services. This shall also apply to nonconforming products and services detected after delivery of products, during or after the provision of services.
组织应根据不合格项的性质及其对产品不符合要求的影响采取适当措施。这也适用于在产品及服务提供期间或之后发现的不合格项。
ISO 13485:2016—Medical devices — Quality management systems — Requirements for regulatory purposes 医疗器械—质量管理体系—监管目的要求
8.3.1 General -- The organization shall ensure that product which does not conform to product requirements is identified and controlled to prevent its unintended use or delivery. The organization shall document a procedure to define the controls and related responsibilities and authorities for the identification, documentation, segregation, evaluation and disposition of nonconforming product.
8.3.1 总则—组织应确保识别和控制不符合要求的产品,以防止其被非预期使用或交付。组织应将其对不合格产品的控制、识别、记录、隔离、评价和处置等做出的规定以及相关职责、权限程序一并形成文件。
The evaluation of nonconformity shall include a determination of the need for an investigation and notification of any external party responsible for the nonconformity.
对不合格产品的评价应包括确定是否需要进行调查和通知负责此不合格产品的任何外部方(external party)。
21 CFR 211— Current Good Manufacturing Practice Finished Pharmaceuticals 制药企业CGMP
Sec. 211.100 Written procedures; deviations. 211.100 书面程序;偏差
(b) Written production and process control procedures shall be followed in the execution of the various production and process control functions and shall be documented at the time of performance. Any deviation from the written procedures shall be recorded and justified.
(b)在执行各种生产和过程控制功能时,应遵循书面生产和工艺控制程序,并在执行过程中以文件进行记录。任何偏离书面程序的情况都应当记录并说明理由。
21 CFR 820— Quality System Regulation 质量体系规范
820.90 Nonconforming product. 820.90 不合格产品
(a) Control of nonconforming product. Each manufacturer shall establish and maintain procedures to control product that does not conform to specified requirements. The procedures shall address the identification, documentation, evaluation, segregation, and disposition of nonconforming product. The evaluation of nonconformance shall include a determination of the need for an investigation and notification of the persons or organizations responsible for the nonconformance. The evaluation and any investigation shall be documented.
(a)不合格产品的控制 每个制造商应建立并维持相应程序来控制不符合规定要求的产品。程序应涉及对不合格产品的识别、记录、评价、隔离和处置。对不合格产品的评价应包括确定是否有必要进行调查或告知负责不合格产品的组织或人员。评价和调查应以文件形式记录。
(b) Nonconformity review and disposition. 不合格产品的审查和处置
(1) Each manufacturer shall establish and maintain procedures that define the responsibility for review and the authority for the disposition of nonconforming product. The procedures shall set forth the review and disposition process. Disposition of nonconforming product shall be documented. Documentation shall include the justification for use of nonconforming product and the signature of the individual(s) authorizing the use.
(1)每个制造商应建立和维持关于不合格产品审查责任和处置权限的程序。程序中应阐明审评和处置不合格产品的过程。对不合格产品的处置应记录形成文件。文件应包括对使用不合格产品的合理解释以及授权使用不合格产品的人员的签字。
GHTF.SG3.N99-8 Guidance on Quality Systems for the Design and Manufacture of Medical Devices
GHTF(The Global Harmonization Task Force,全球协调工作组).SG3.N99-8—医疗器械设计和制造质量体系指南
4.13.1 General 总则
When any intermediate or final product (including service) is found (e.g., by test or inspection) not to conform to the technical specifications, inadvertent use or installation should be prevented. This is applicable to nonconforming product occurring in the supplier's own production as well as nonconforming product received by the supplier.
当发现任何中间产品或最终产品(包括服务)(例如通过试验或检查发现)不符合技术规范时,应当防止其被非预期或意外使用或安装。该规定适用于供应商在生产中发现不合格产品以及收到不合格产品。
An important element in addressing nonconformities is to give to all appropriate personnel the freedom to identify nonconforming items, activities and processes and encouragement to suggest improvements.
为解决不合格问题,企业应当保证所有的相关人员可自由识别不合格项目、活动和过程,并鼓励他们提出改进意见。
ICH Harmonized Tripartite Guideline Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients Q7
ICH协调三方指导原则—Q7 原料药优良制造规范
2.1 Principles 原则
2.16 Any deviation from established procedures should be documented and explained. Critical deviations should be investigated, and the investigation and its conclusions should be documented.
2.16 任何与既定程序的偏离都应该记录并解释。应调查严重偏差,调查过程及结论应该记录形成文件。
ICH Harmonized Tripartite Guideline Pharmaceutical Quality System Q10 ICH 协调三方指导原则—Q10 药品质量体系
3.2.1 Process Performance and Product Quality Monitoring System 过程绩效与产品质量监控体系
(e) Include feedback on product quality from both internal and external sources, e.g., complaints, product rejections, nonconformances, recalls, deviations, audits, and regulatory inspections and findings.
(e)包括来自内部和外部的产品质量反馈,例如投诉、产品退回(product rejections)、不合格、召回、偏差、审核及监管检查和调查结果。
The above regulations, standards, and guidance documents either refer directly or indirectly to the use of risk-based thinking to manage nonconformances/deviations.
上述法规、标准及指南文件或直接或间接地使用基于风险的思维来管理不合格及偏差现象。
二、Nonconformance And Deviation Classification
不合格和偏差分类
Risk-based thinking should be an integral part of an effective and efficient nonconformance and deviation management program. The level of control should be proportionate to the effect on the quality of the product produced or services provided by your organization. It should be obvious that as the risk level of the nonconformance and deviation increases, so should the requirements and controls used to manage nonconformances and deviations.
基于风险的思维是不合格和偏差管理计划的重要组成部分。风险控制水平应与风险对组织所生产产品或所提供服务质量的影响相当。很显然,随着不合格和偏差的风险水平提高,用于管理不合格和偏差的要求和控制也应该增加。
Table 1 provides example definitions for low-, medium-, and high-impact nonconformances and deviations. Once the risk level has been determined (low, medium, or high), the appropriate risk-based nonconformance and deviation controls can be applied.
表1中提供了关于不合格和偏差低、中、高影响的的定义。一旦确定了风险等级(低、中或高),就可以应用相应的基于风险的不合格和偏差控制方法对风险进行管理。
表1 影响定义,风险可接受性和控制要求

三、Nonconformance And Deviation Management
不合格和偏差管理
There are generally two methods to manage nonconformances and deviations. The first is through the nonconformances and deviations process; the second is the corrective and preventive action (CAPA) process. The CAPA process is primarily used for high- and medium-risk issues, while the nonconformances and deviations process is used for medium- and low-risk issues.
企业通常会通过两种方法来管理不合格和偏差。一是不合格和偏差过程;二是纠正和预防措施(CAPA)过程。CAPA过程主要用于高风险和中等风险问题,而不合格和偏差过程则用于中等和低风险问题。
表2 典型的纠正和预防措施过程步骤

The CAPA process has eight distinct steps or phases, including problem identification, impact assessment, remedial action/containment, investigation/root cause analysis, corrective action, implementation, verification of effectiveness, and closure. Each step has specific requirements that should be followed to ensure successful resolution of quality issues, including:
CAPA过程有8个不同的步骤或阶段,包括问题识别、影响评估、补救措施/遏制、调查/根本原因分析、纠正措施、实施、有效性验证和关闭。每一步都有具体的要求,确保能够成功和有效解决质量问题,具体包括:
Problem identification – describe the problem and its source
问题识别—描述问题及其来源
Impact assessment – what products, processes, or systems may be affected
影响评估—可能会影响哪些产品、过程或系统
Remedial action/containment – place product on hold, recall, quarantine, etc.
补救措施/遏制—将产品搁置、召回、隔离等
Investigation/root cause analysis – determine what caused the issue
调查/根本原因分析—确定引起的问题的原因
Corrective action – actions taken to address the root cause of the problem
纠正措施—为解决问题的根源而采取的措施
Implementation – the deployment of corrective action(s)
实施—纠正措施的部署及执行
Verification of effectiveness – the plan, criteria, and requirements to ensure the problem will not recur or have other adverse effects.
有效性验证—确保问题不再发生,或实施的计划、标准和要求不再具有其他不利影响
Closure – ensuring the corrective action(s) were effective; this should include a disposition record of any products or materials affected.
关闭—确保纠正措施是有效的,包括任何受影响的产品或材料的处置记录
表3 典型的不合格和偏差过程步骤
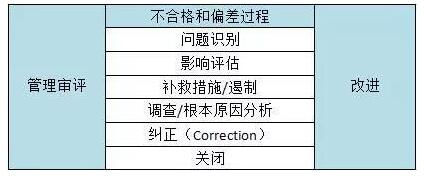
The nonconformances and deviations process has six steps or phases, including problem identification, impact assessment, remedial action/containment, investigation/root cause analysis, correction, and closure. Each step has distinct requirements that should be followed to ensure successful resolution of quality issues, including:
不合格和偏差过程分为6个步骤或阶段,包括:问题识别、影响评估、补救措施/遏制、调查/根本原因分析、纠正和关闭。每一步都有不同的要求,应该遵循以下规定以确保能够成功解决质量问题,包括:
Problem identification – describe the problem and its source
问题识别—描述问题及其来源
Impact assessment – what products, processes, or systems may be affected
影响评估—可能会影响哪些产品、过程或系统
Remedial action/containment – place product on hold, recall, quarantine, etc.
补救措施/遏制—将产品搁置、召回、隔离等
Investigation/root cause analysis – determine what caused the issue
调查/根本原因分析—确定引起的问题原因
Correction – actions taken to address the root cause of the problem
纠正—为解决问题的根源而采取的措施
Closure – ensuring the correction was completed; this should include a disposition record of any products or materials affected
关闭—确保纠正措施是有效的,包括任何受影响的产品或材料的处置记录
The CAPA process and the nonconformances and deviations process are very similar, except for corrective action vs. correction, implementation, and verification of effectiveness. To better understand these differences, Govind Ramu defines the difference as: “Correction is an action taken to eliminate a detected nonconformity,” and “Corrective action is taken to eliminate the cause of a detected nonconformity.” Ramu further states, “Both correction and corrective action may be required in many scenarios. Correction addresses the short-term need and gets immediate attention, and most organizations do a good job of correcting the nonconformity. Corrective action, on the other hand, is a long-term solution … organizations do not invest adequate resources in addressing corrective action.”Due to the short-term nature of corrections, the implementation and verification of effectiveness phases are generally not required or completed by most organizations.
除了“纠正措施”(Corrective action)与“纠正”(Correction)、“实施”和“有效性验证”步骤不同之外,CAPA过程与不合格和偏差过程非常相似。为了更好地理解这些差异,可进行以下定义:“纠正”是消除检测到的不合格或偏差而采取的措施,而“纠正措施”则是用于消除产生不合格或偏差的原因。许多情况下可能同时需要纠正及纠正措施来预防风险。“纠正”通常用于解决短期需要,并可以得到立竿见影的效果,大部分组织可通过纠正管理不合格的工作。由于其短期性质,大多数组织通常不需要或完成“实施”和“有效性验证”阶段。相反,“纠正措施”是一个长期的解决方案,但是组织一般不会投入大量的资源启动“纠正措施”。
四、Conclusion 结论
The discussion above shows various opportunities for integrating risk management concepts to manage nonconformances and deviations. The concepts presented can be readily applied to virtually any industry as best practices.
以上讨论通过整合风险管理理念对企业的不合格和偏差管理进行分析。本文中提出的定义和要求可以而且应该根据组织的风险程度、接受阈值、行业实践、指南文件和法规要求酌情应用,并且企业可探索符合自身情况的最佳实践。
全部 0条评论