
1 回答

在2021年1月12日国家局药监局发布的《药品上市后变更管理办法(试行)》的公告(2021年第8号)中,第七条,第八条明确规定了国内持有人主体变更的程序和需要开展的工作。
《药品上市后变更管理办法(试行)》的公告(2021年第8号)第七条 申请变更药品持有人的,药品的生产场地、处方、生产工艺、质量标准等应当与原药品一致;发生变更的,可在持有人变更获得批准后,由变更后的持有人进行充分研究、评估和必要的验证,并按规定经批准、备案后实施或报告。
【分析:这里就明确说了,申请变更药品持有人的,生产场地,处方,生产工艺,质量标准等时,是先进行持有人变更,持有人变更获得批准后,由变更后的持有人进行充分研究,评估和必要的验证,并按规定经批准、备案后实施或报告;这样也是可以节省持有人的变更的成本,若是反过来的话,先变更场地等,得先进行三批工艺验证,进行变更场地前后的质量可比性研究,通过后,再进行持有人变更,变更持有人的时候,新的持有人需要取得B证,可能还需要进行工艺验证,然后再获批,这样这样持有人可能进行两次工艺验证,耗时耗力耗财】
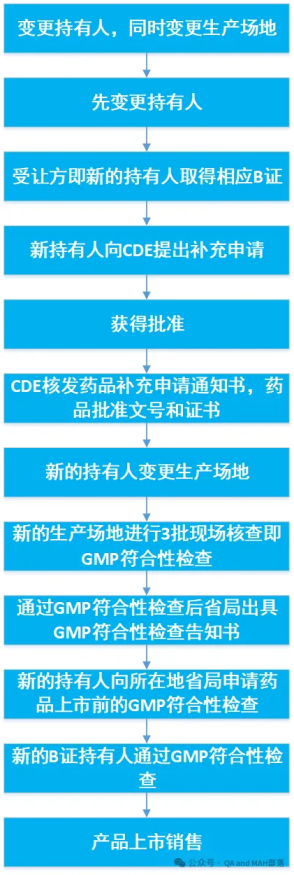
此处是以变更持有人,同时变更生产场地为例做的流程图,若是变更持有人同时变更生产工艺,质量标准等,流程类似。
《药品上市后变更管理办法(试行)》的公告(2021年第8号)第八条 申请变更境内生产药品的持有人,受让方应当在取得相应生产范围的药品生产许可证后,向国家药品监督管理局药品审评中心(以下简称药审中心)提出补充申请。其中,申请变更麻醉药品和精神药品的持有人,受让方还应当符合国家药品监督管理局确定的麻醉药品和精神药品定点生产企业的数量和布局要求。
药审中心应当在规定时限内作出是否同意变更的决定,同意变更的,核发药品补充申请通知书,药品批准文号和证书有效期不变,并抄送转让方、受让方和生产企业所在地省级药品监管部门。
变更后的持有人应当具备符合药品生产质量管理规范要求的生产质量管理体系,承担药品全生命周期管理义务,完成该药品的持续研究工作,确保药品生产上市后符合现行技术要求,并在首次年度报告中重点说明转让的药品情况。
转让的药品在通过药品生产质量管理规范符合性检查后,符合产品放行要求的,可以上市销售。
受让方所在地省级药品监管部门应当重点加强对转让药品的监督检查,及时纳入日常监管计划。
这家伙很懒,还没有设置简介